# gtfsort
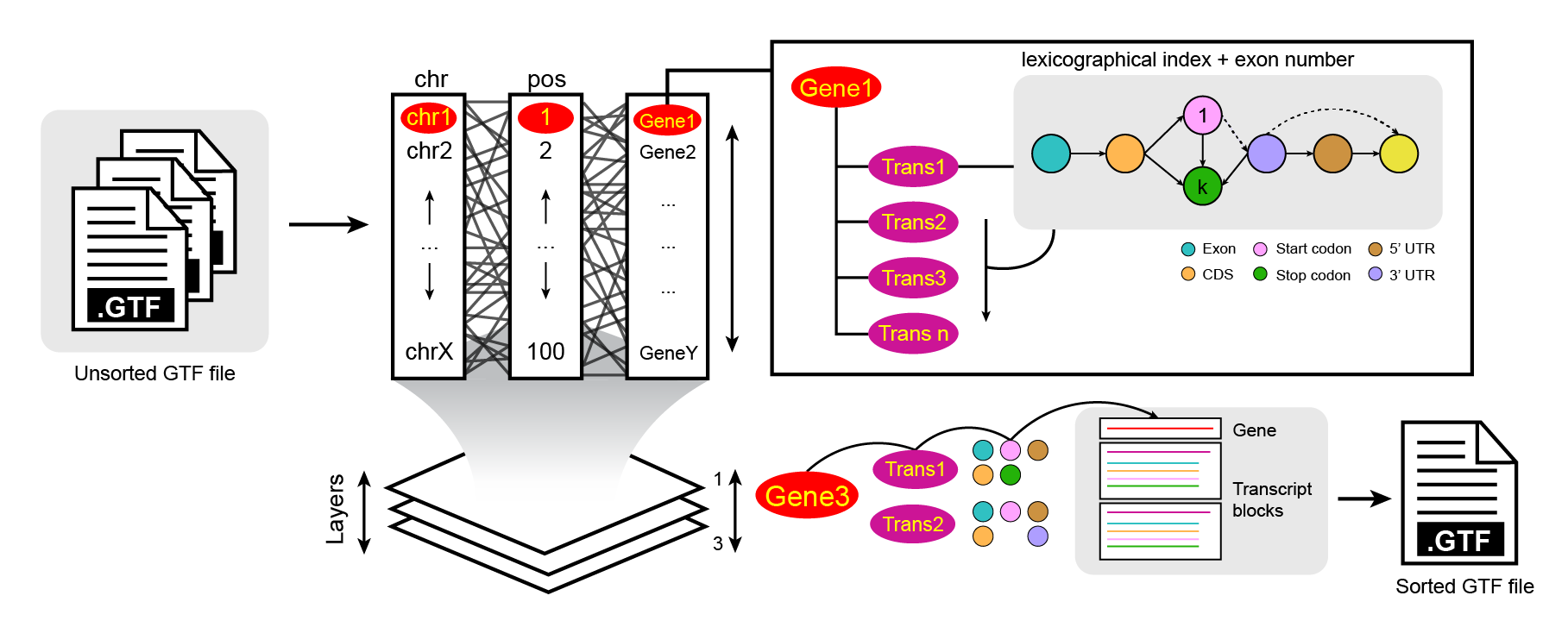
An optimized chr/pos/feature GTF/GFF sorter using a lexicographically-based index ordering algorithm written in Rust.
> - Now supporting GFF files!
While current tools (most of them GFF3-focused) have been recommended for sorting GTF files, none are directed towards chr/pos/feature ordering. This approach ensures custom sorting directionality, which is useful for reducing computation times in tools that work with sorted GTF files. Furthermore, it provides a friendly and organized visualization of gene structures (gene -> transcript -> CDS/exon -> start/stop -> UTR/Sel), allowing users to search for features more efficiently.
>[!NOTE]
>
> If you use gtfsort in your work, please cite:
>
> Gonzales-Irribarren A. gtfsort: a tool to efficiently sort GTF files. bioRxiv 2023.10.21.563454; doi: https://doi.org/10.1101/2023.10.21.563454
## Usage
``` rust
Usage: gtfsort -i -o