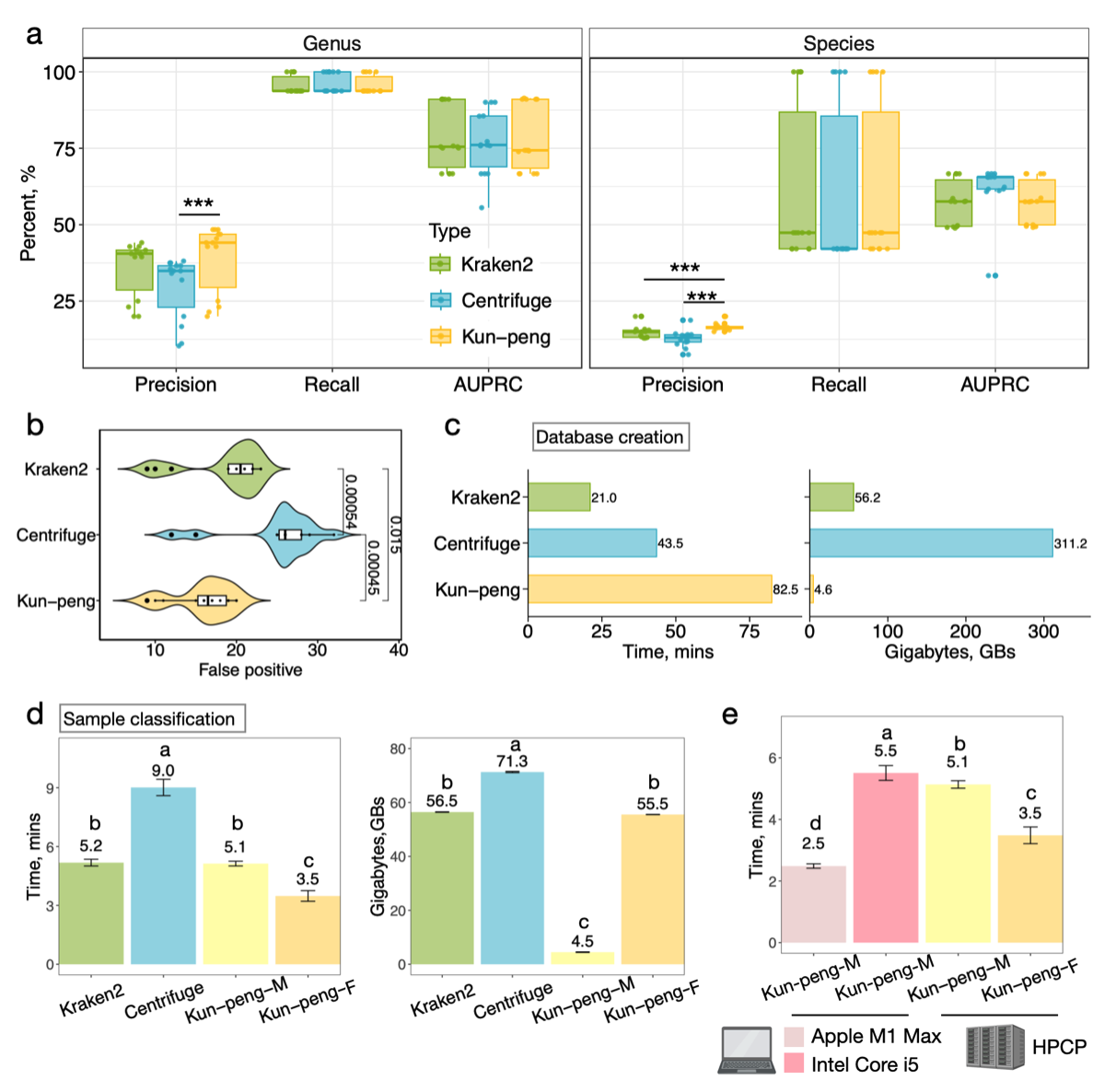
Fig. 2. Performance benchmark of Kun-peng against other metagenomic classifiers.
References:
1. Lu, J., Breitwieser, F. P., Thielen, P. & Salzberg, S. L. Bracken: Estimating species abundance in metagenomics data. PeerJ Comput. Sci. 2017, 1–17 (2017).
2. Amos, G. C. A. et al. Developing standards for the microbiome field. Microbiome 8, 1–13 (2020).
3. Kralj, J., Vallone, P., Kralj, J., Hunter, M. & Jackson, S. Reference Material 8376 Microbial Pathogen DNA Standards for Detection and Identification NIST Special Publication 260-225 Reference Material 8376 Microbial Pathogen DNA Standards for Detection and Identification.
4. Wood, D. E., Lu, J. & Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 20, (2019).
5. Kim, D., Song, L., Breitwieser, F. P. & Salzberg, S. L. Centrifuge: Rapid and sensitive classification of metagenomic sequences. Genome Res. 26, 1721–1729 (2016).
6. Ye, S. H., Siddle, K. J., Park, D. J. & Sabeti, P. C. Benchmarking Metagenomics Tools for Taxonomic Classification. Cell 178, 779–794 (2019).
## Get Started
Follow these steps to install Kun-peng and run the examples.
### Method 1: Download Pre-built Binaries (Recommended)
If you prefer not to build from source, you can download the pre-built binaries for your platform from the GitHub [releases page](https://github.com/eric9n/Kun-peng/releases).
For Linux users (CentOS 7 compatible):
```bash
# Replace X.Y.Z with the latest version number
VERSION=vX.Y.Z
mkdir kun_peng_$VERSION
wget https://github.com/eric9n/Kun-peng/releases/download/$VERSION/kun_peng-$VERSION-centos7
# For linux x86_64
# wget https://github.com/eric9n/Kun-peng/releases/download/$VERSION/kun_peng-$VERSION-x86_64-unknown-linux-gnu
mv kun_peng-$VERSION-centos7 kun_peng_$VERSION/kun_peng
chmod +x kun_peng_$VERSION/kun_peng
# Add to PATH
echo "export PATH=\$PATH:$PWD/kun_peng_$VERSION" >> ~/.bashrc
source ~/.bashrc
```
For macOS users:
### Homebrew
```bash
brew install eric9n/tap/kun_peng
```
### Binary
```bash
# Replace X.Y.Z with the latest version number
VERSION=vX.Y.Z
mkdir kun_peng_$VERSION
# For Intel Macs
wget https://github.com/eric9n/Kun-peng/releases/download/$VERSION/kun_peng-$VERSION-x86_64-apple-darwin
mv kun_peng-$VERSION-x86_64-apple-darwin kun_peng_$VERSION/kun_peng
# For Apple Silicon Macs
# wget https://github.com/eric9n/Kun-peng/releases/download/$VERSION/kun_peng-$VERSION-aarch64-apple-darwin
# mv kun_peng-$VERSION-aarch64-apple-darwin kun_peng_$VERSION/kun_peng
chmod +x kun_peng_$VERSION/kun_peng
# Add to PATH
echo "export PATH=\$PATH:$PWD/kun_peng_$VERSION" >> ~/.zshrc # or ~/.bash_profile for Bash
source ~/.zshrc # or source ~/.bash_profile for Bash
```
For Windows users:
```powershell
# Replace X.Y.Z with the latest version number
$VERSION = "vX.Y.Z"
New-Item -ItemType Directory -Force -Path kun_peng_$VERSION
Invoke-WebRequest -Uri "https://github.com/eric9n/Kun-peng/releases/download/$VERSION/kun_peng-$VERSION-x86_64-pc-windows-msvc.exe" -OutFile "kun_peng_$VERSION\kun_peng.exe"
# Add to PATH
$env:Path += ";$PWD\kun_peng_$VERSION"
[Environment]::SetEnvironmentVariable("Path", $env:Path, [EnvironmentVariableTarget]::User)
```
After installation, you can verify the installation by running:
```bash
kun_peng --version
```
#### Run the `kun_peng` example
We will use a very small virus database on the GitHub homepage as an example:
1. clone the repository
``` sh
git clone https://github.com/eric9n/Kun-peng.git
cd Kun-peng
```
2. build database
``` sh
kun_peng build --download-dir data/ --db test_database
```
```
merge fna start...
merge fna took: 29.998258ms
estimate start...
estimate count: 14080, required capacity: 31818.0, Estimated hash table requirement: 124.29KB
convert fna file "test_database/library.fna"
process chunk file 1/1: duration: 29.326627ms
build k2 db took: 30.847894ms
```
3. classify
``` sh
# temp_chunk is used to store intermediate files
mkdir temp_chunk
# test_out is used to store output files
mkdir test_out
kun_peng classify --db test_database --chunk-dir temp_chunk --output-dir test_out data/COVID_19.fa
```
```
hash_config HashConfig { value_mask: 31, value_bits: 5, capacity: 31818, size: 13051, hash_capacity: 1073741824 }
splitr start...
splitr took: 18.212452ms
annotate start...
chunk_file "temp_chunk/sample_1.k2"
load table took: 548.911µs
annotate took: 12.006329ms
resolve start...
resolve took: 39.571515ms
Classify took: 92.519365ms
```
### Method 2: Clone the Repository and Build the project
#### Prerequisites
1. **Rust**: This project requires the Rust programming environment if you plan to build from source.
#### Build the Projects
First, clone this repository to your local machine:
``` sh
git clone https://github.com/eric9n/Kun-peng.git
cd kun_peng
```
Ensure that both projects are built. You can do this by running the following command from the root of the workspace:
``` sh
cargo build --release
```
This will build the kr2r and ncbi project in release mode.
#### Run the `kun_peng` example
Next, run the example script that demonstrates how to use the `kun_peng` binary. Execute the following command from the root of the workspace:
``` sh
cargo run --release --example build_and_classify
```
This will run the build_and_classify.rs example located in the kr2r project's examples directory.
Example Output You should see output similar to the following:
``` txt
Executing command: /path/to/workspace/target/release/kun_peng build --download-dir data/ --db test_database
kun_peng build output: [build output here]
kun_peng build error: [any build errors here]
Executing command: /path/to/workspace/target/release/kun_peng direct --db test_database data/COVID_19.fa
kun_peng direct output: [direct output here]
kun_peng direct error: [any direct errors here]
```
This output confirms that the `kun_peng` commands were executed successfully and the files were processed as expected.
## ncbi_dl tool
For detailed information and usage instructions for the ncbi_dl tool, please refer to the [ncbi_dl repository](https://github.com/eric9n/ncbi_dl.git).
The ncbi_dl tool is used to download resources from the NCBI website, including taxonomy files and genome data. It provides a convenient way to obtain the necessary data for building Kun-peng databases.
### Downloading Genome Databases
To download genome databases using ncbi_dl, you can use the `genomes` (or `gen`) command. Here's a basic example:
```sh
ncbi_dl -d /path/to/download/directory gen -g bacteria
```
This command will download bacterial genomes to the specified directory. You can replace `bacteria` with other genome groups like `archaea`, `fungi`, `protozoa`, or `viral` depending on your needs.
Some key options for the `genomes` command include:
- `-g, --groups 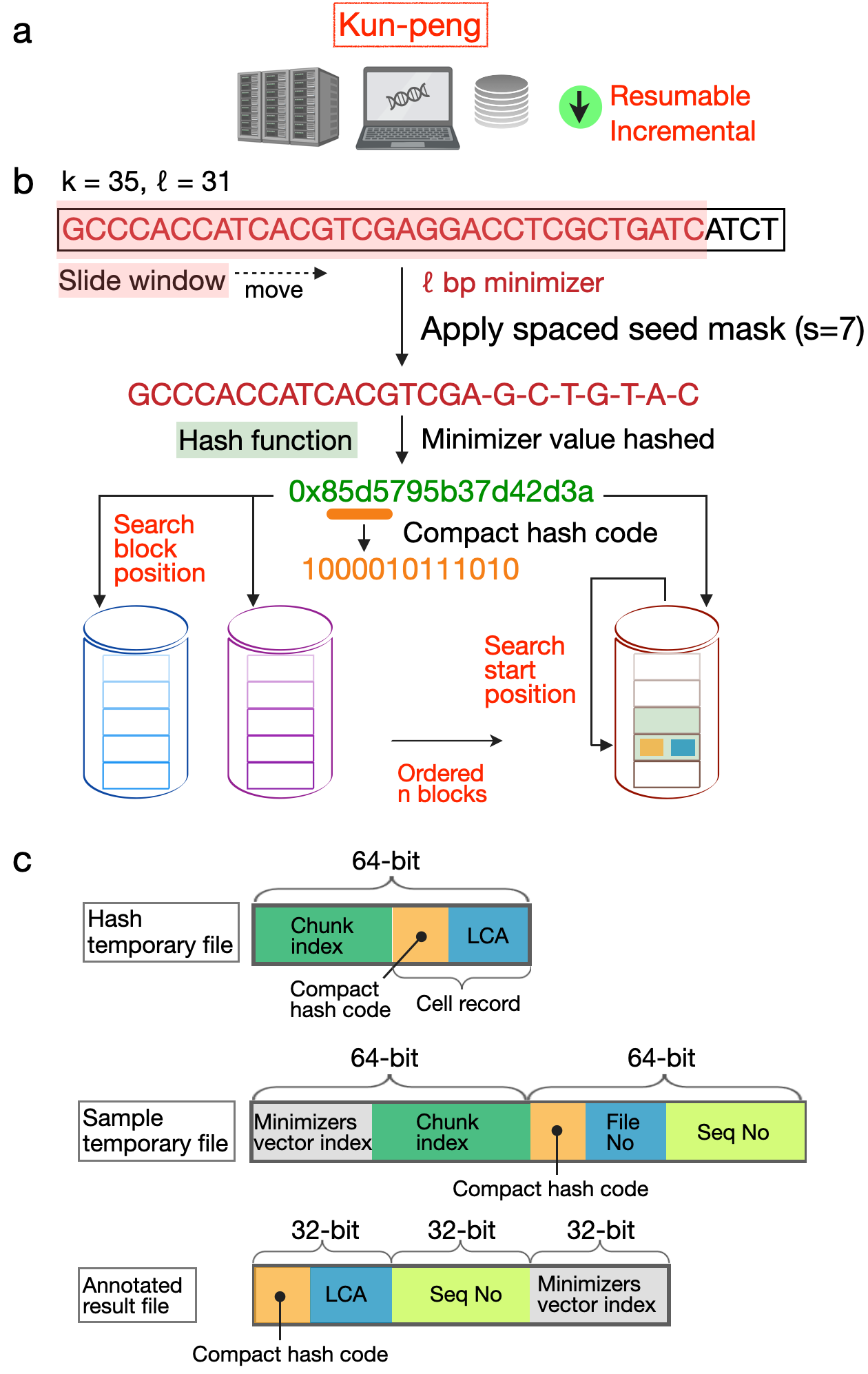
 []() [](https://github.com/eric9n/Kun-peng/releases)
Here, we introduce Kun-peng, an ultra-memory-efficient metagenomic classification tool (Fig. 1). Inspired by Kraken2's k-mer-based approach, Kun-peng employs algorithms for minimizer generation, hash table querying, and classification. The cornerstone of Kun-peng's memory efficiency lies in its unique ordered block design for reference database. This strategy dramatically reduces memory usage without compromising speed, enabling Kun-peng to be executed on both personal computers and HPCP for most databases. Moreover, Kun-peng incorporates an advanced sliding window algorithm for sequence classifications to reduce the false-positive rates. Finally, Kun-peng supports parallel processing algorithms to further bolster its speed. Kun-peng offers two classification modes: Memory-Efficient Mode (Kun-peng-M) and Full-Speed Mode (Kun-peng-F). Remarkably, Kun-peng-M achieves a comparable processing time to Kraken2 while using less than 10% of its memory. Kun-peng-F loads all the database blocks simultaneously, matching Kraken2's memory usage while surpassing its speed. Notably, Kun-peng is compatible with the reference database built by Kraken2 and the associated abundance estimate tool Bracken1, making the transition from Kraken2 effortless. The name "Kun-peng" was derived from Chinese mythology and refers to a creature transforming between a giant fish (Kun) and a giant bird (Peng), reflecting the software's flexibility in navigating complex metagenomic data landscapes.
[]() [](https://github.com/eric9n/Kun-peng/releases)
Here, we introduce Kun-peng, an ultra-memory-efficient metagenomic classification tool (Fig. 1). Inspired by Kraken2's k-mer-based approach, Kun-peng employs algorithms for minimizer generation, hash table querying, and classification. The cornerstone of Kun-peng's memory efficiency lies in its unique ordered block design for reference database. This strategy dramatically reduces memory usage without compromising speed, enabling Kun-peng to be executed on both personal computers and HPCP for most databases. Moreover, Kun-peng incorporates an advanced sliding window algorithm for sequence classifications to reduce the false-positive rates. Finally, Kun-peng supports parallel processing algorithms to further bolster its speed. Kun-peng offers two classification modes: Memory-Efficient Mode (Kun-peng-M) and Full-Speed Mode (Kun-peng-F). Remarkably, Kun-peng-M achieves a comparable processing time to Kraken2 while using less than 10% of its memory. Kun-peng-F loads all the database blocks simultaneously, matching Kraken2's memory usage while surpassing its speed. Notably, Kun-peng is compatible with the reference database built by Kraken2 and the associated abundance estimate tool Bracken1, making the transition from Kraken2 effortless. The name "Kun-peng" was derived from Chinese mythology and refers to a creature transforming between a giant fish (Kun) and a giant bird (Peng), reflecting the software's flexibility in navigating complex metagenomic data landscapes.