genovo
| Crates.io | genovo |
| lib.rs | genovo |
| version | 0.3.2 |
| created_at | 2020-08-07 10:29:15.242912+00 |
| updated_at | 2022-09-16 09:52:54.771541+00 |
| description | Determine genes with significantly more mutations than expected by chance |
| homepage | |
| repository | https://github.com/BesenbacherLab/genovo |
| max_upload_size | |
| id | 273932 |
| size | 9,708,702 |
 (besenbacher)
(besenbacher)
documentation
README
GeNovo - Identifying disease genes with de-novo mutations
Everyone's genome contains some de-novo mutations: Mutations that are not part of their parents' genomes. These de-novo mutations can cause diseases but it is still difficult to tell the disease-causing mutations apart from the ones that have nothing to do with the disease. Furthermore, some genes have a higher chance of mutating which means that a mutation in that gene is not that surprising, even when that gene is mutated in several people with the disease.
The name GeNovo is a wordplay of gene detection based on de-novo mutations.
Installation
First make sure you have cargo installed (cargo is the package manager for the Rust programming language).
The easiest way is to install rustup which automatically installs cargo and rustc for you.
If you are on a UNIX-based system, you might also be able to install cargo or rustup with your system's package manager.
Secondly, you need to run
cargo install genovo
This will install the program genovo at a specific location which will be shown at the end of the installation process.
(You might need to make adjustments to your PATH variable afterwards to automatically find the program on the command line)
The pipeline is a single executable (genovo) that combines the important steps from an earlier pipeline that I had put together.
(A single executable is much easier for others to use than a bunch of scripts that are all loosely coupled.)
The pipeline
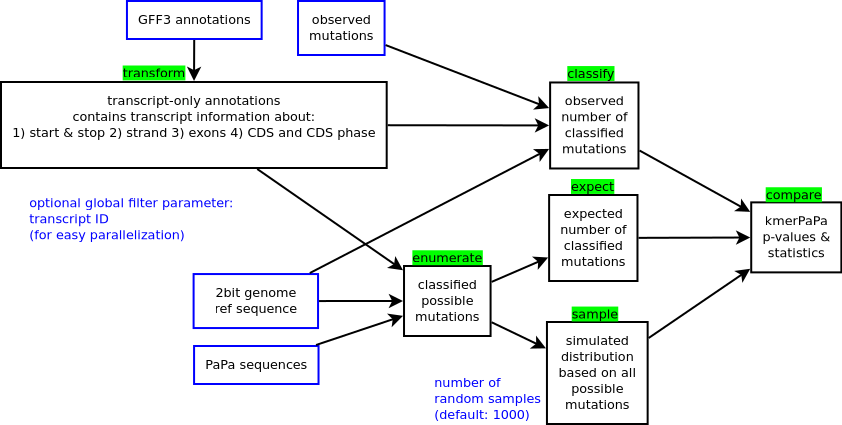
- Input files are marked with a blue border.
- Pipeline steps are marked with a black border and their names are marked with a green background
- Optional parameters are marked in blue font
Input and output files
The pipeline takes several parameters that represent input and output parameters. Depending on which action is being performed, a parameter is treated either as an input or output file.
For example, the --possible-mutations parameter is treated as an input file when --action expect is specified.
But it is treated as an output file when --action enumerate is specified.
If no --action parameter is specified, the entire pipeline is run and all file parameters are treated as output files.
You can also specify - as an input/output file. Depending on the action, this will be interpreted as STDIN or as STDOUT.
If you are concerned about space usage you can also specify a .gz file extension.
genovo will automatically decompress/compress the input/output.
ID filtering / parallel processing
If the parameter --id ID is passed, then only the transcripts with the id ID will be processed
and all other transcripts will be ignored.
This is useful for parallel execution. You can launch a separate job for each transcript.
Please note that --id only accepts a single ID. Multiple IDs are not supported.
You can always combine the --id option with any other option.
Running the whole pipeline on a test data set
You can download a test data set from the git repository:
wget https://github.com/BesenbacherLab/genovo/raw/master/test_data/test_data.tar.gz
tar xvzf test_data.tar.gz
If you want to run the entire pipeline on the test data you can execute the following command:
genovo \
--gff3 gencode.v19.CCDS.annotation.protein_coding_chr22.gff3.gz \
--observed-mutations observed_mutations_DDD_2017_chr22.txt \
--genome hg19_chr22.2bit \
--point-mutation-probabilities SNV_mutation_rate_model.txt \
--indel-mutation-probabilities indel_mutation_rate_model.txt \
--scaling-factor 8586 \
--significant-mutations genovo_results_chr22.txt
The command takes ~1 minute to run on a "normal" computer.
It will write the comparisons between observed, expected and sampled mutations to the file genovo_results_chr22.txt. If the --significant-mutations argument is not used the results will be written to STDOUT.
For each transcript, t, and mutation type, m, the output contains the following output columns
| column | description |
|---|---|
| observed | The observed number of m mutations in transcript t |
| expected | The expected number of m mutations in transcript t according to the mutation rate models |
| expected_lower | The lower bound of the confidence interval around the expected number of mutations |
| expected_upper | The upper bound of the confidence interval around the expected number of mutations |
| p_value | A one-sided p-value for a sampling based test of whether there are more observed mutations of type m in transcript t than we would expect given our mutation rate models |
When testing on de novo mutations from trios the --scaling-factor should be two times the number of trios in the data. So in this case it is set to 8586 since the data set with the observed mutations looked for de novo mutations in 4293 children.
The SNV_mutation_rate_model.txt and indel_mutation_rate_model.txt are mutation rate models created using kmerpapa. Trained models in the right input format can be downloaded from https://github.com/BesenbacherLab/Genovo_Input.
Steps of the pipeline
Each of the steps of the pipeline can be executed in isolation by specifying the --action STEP parameter.
If no --action parameter is specified, all steps are run (as in the example above).
transform
This step takes a gff3 file and transforms it into a regions file (which is a file format that is specific to genovo).
The gff3 file needs to have the following attributes:
- ID: For identifying the name of each transcript
- Parent: For identifying the parent of exons and CDS regions
The file must also be sorted so that all exons and CDS regions are listed after their transcript line.
Some example gff3 files can be obtained from GENCODE
It is highly recommended that you only use annotations that contain exclusively coding transcripts. The results for non-coding transcripts are a lot less informative.
parameters
| parameter | input/output | description |
|---|---|---|
| --gff3 | input | The GFF3 annotation file that contains transcript, exon and CDS information |
| --genomic-regions | output | A transformed version of the GFF3 entries, limited to transcripts, exons and CDS regions |
enumerate
This goes through the genomic sequence of every transcript and determines all possible point mutations together with their probability This command takes a PaPa-rates file which represents the point mutation probabilities for different sequence contexts as described in our paper *Improved prediction of site-specific mutation rates using k-mer pattern partition *. This command also takes a reference genome file in the twobit file format. Make sure that all your input files use the same genome build!
The output file is fairly simple:
#transcript_ID
numeric_mutation_type:mutation_probability
numeric_mutation_type:mutation_probability
numeric_mutation_type:mutation_probability
#other_transcript_ID
numeric_mutation_type:mutation_probability
numeric_mutation_type:mutation_probability
numeric_mutation_type:mutation_probability
numeric_mutation_type:mutation_probability
numeric_mutation_type:mutation_probability
numeric_mutation_type:mutation_probability
numeric_mutation_type:mutation_probability
The possible mutations for each transcript start with a #ID line followed
with one line for every possible mutation where the probability is not NaN.
Keep in mind that every position may mutate into one of the 3 other bases, so
that you have 3 possible point mutations for every position.
The numeric mutation type is defined in the section Mutation type numeric code table.
parameters
| parameter | input/output | description |
|---|---|---|
| --genomic-regions | input | A transformed version of the GFF3 entries, limited to transcripts, exons and CDS regions |
| --genome | input | A twobit reference genome sequence file |
| --point-mutation-probabilities | input | A pattern partition file with the sequence-context-dependent point mutation probabilities |
| --indel-mutation-probabilities | input | A pattern partition file with the sequence-context-dependent indel probabilities |
| --possible-mutations | output | The possible mutations for each transcript |
classify
This step classifies observed point mutations (which is a raw input file you should provide). The input file format is as follows:
chr1 1230448 G A
chr1 1609723 C T
chr1 1903276 C T
chr1 2574999 C T
The first column is the chromosome (must be the same naming convention as in the annotations and the twobit file). The second column is the position on the chromosome. This position has to be 1-based. The third column is the reference base. If this reference base does not match what is in the twobit file, you will get an error. The fourth column is the new base that was mutated into.
parameters
| parameter | input/output | description |
|---|---|---|
| --observed-mutations | input | Observed point mutations from your own data |
| --genome | input | A twobit reference genome sequence file |
| --genomic-regions | input | A transformed version of the GFF3 entries, limited to transcripts, exons and CDS regions |
| --classified-mutations | output | The observed mutatations. But annotated with numeric mutation types |
expect
This step sums up all possible mutations and their probabilities and gives you a grand total that represents the expected number of mutations for each gene.
parameters
| parameter | input/output | description |
|---|---|---|
| --possible-mutations | input | The possible mutations for each transcript |
| --expected-mutations | output | The number of expected mutations per transcript for each mutation type |
sample
This step randomly picks possible mutations and simulates randomly if they occur based on their probability. This creates an empirical distribution of sampled mutations that will be used to calculate a p-value in a later step.
parameters
| parameter | input/output | description |
|---|---|---|
| --possible-mutations | input | The possible mutations for each transcript |
| --number-of-random-samples | integer | How many random samples? default=1000 |
| --sampled-mutations | output | Distributions of sampled mutations |
compare
Compare the observed, expected and sampled mutations to determine a p-value for observing that many mutations in a transcript by chance. Please note that the output file contains the results of all statistical tests, even the non-significant ones. And note that the p-values does not take into account the expected number of mutations.
parameters
| parameter | input/output | description |
|---|---|---|
| --classified-mutations | input | The observed mutatations. But annotated with numeric mutation types |
| --expected-mutations | input | The number of expected mutations per transcript for each mutation type |
| --sampled-mutations | input | Distributions of sampled mutations |
| --significant-mutations | output | A table of observed, expected, sampled and a p-value for different mutation types. |
Mutation type numeric code table
The input and output files of the pipeline are almost all text files. In order to be more space-efficient, there is a numeric encoding for the different types of mutations.
| mutation type name | numeric code |
|---|---|
| unknown | 0 |
| synonymous | 1 |
| missense | 2 |
| nonsense | 3 |
| loss of stop codon | 4 |
| loss of start codon | 5 |
| loss of canonical splice site | 6 |
| Mutation within an intron | 7 |
| Non-frameshift indel | 8 |
| Frameshift indel | 9 |